
Decoding the Cancer Mechanisms of Fibrolamellar Carcinoma: How DNAJB1-PRKACA drives Cancer Growth
Critical cellular functions such as growth, survival, and communication with other cell types in the body are mediated by networks of proteins. These networks are normally tightly regulated by signaling molecules, which serve as on/off switches. Genetic changes can cause these signaling molecules to become dysregulated, leading to uncontrolled cell growth and cancer development. The hallmark genetic change in fibrolamellar carcinoma (FLC) generates an abnormal fusion of two proteins, DNAJB1 and PRKACA (DNAJB1-PRKACA), which result in the PRKACA portion—a type of signaling protein called a kinase— being constantly switched on.
While medicines that block the function of DNAJB1-PRKACA slow tumors in mouse models of FLC, they also affect PRKACA in normal cells of the body and may not be safe for patients. This study sought to uncover the networks controlled by DNAJB1-PRKACA that lead to cancer. Addressing this question is important since defining how DNAJB1-PRKACA causes cancer growth more precisely could identify other targets for drugs that may be safe and effective.
Using tumor cells derived from patients with FLC as well as mouse cells altered to produce DNAJB1-PRKACA, the research team identified a series of biochemical changes that are required for this cancer-causing function. The cascade of changes begins with DNAJB1-PRKACA directly turning off proteins called Salt-Inducible Kinases (SIKs) through a biochemical modification known as phosphorylation. Normally, SIKs help maintain cellular balance by regulating gene expression (the production of proteins from the genetic code, i.e. DNA) and preventing excessive cell growth. However, in FLC the fusion protein inhibits SIKs, leading to a loss of regulatory control and the activation of cancer-promoting pathways. The researchers found that blocking the function of SIK was sufficient to override the anti-tumor response that came from PRKACA inhibition in FLC models.
Moreover, the study went a step further, determining the key cancer-driving changes resulting from the loss of SIK function. By measuring the levels of thousands of genes, which dictate the properties of FLC cells, the investigators showed that a great many of the genes controlled by DNAJB1-PRKACA were mediated by SIK suppression.
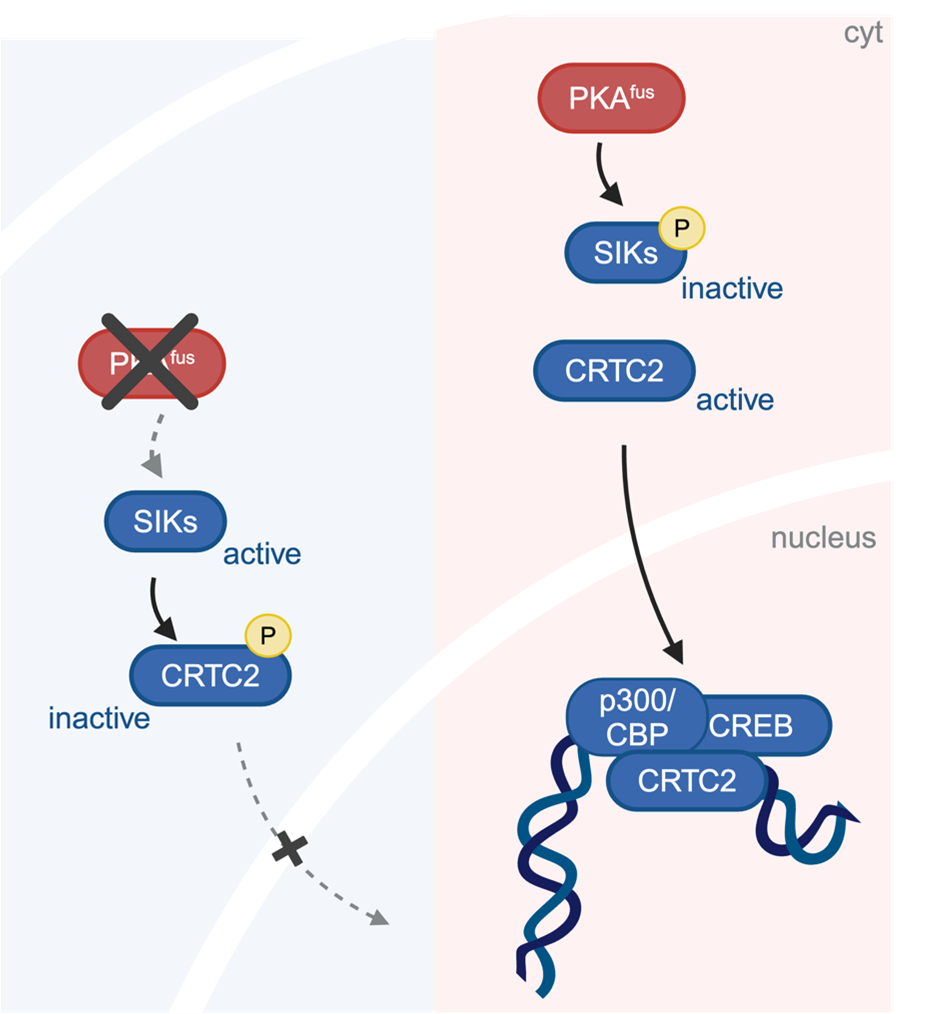
(Created in BioRender. Gritti, I. (2024)
BioRender.com/n45w785)
SIK does not affect genes directly, but affects other proteins that do so, including CRTC2. The investigators found that CRTC2, which is normally phosphorylated and trapped in the cytoplasm of the cell by SIK, is not phosphorylated in FLC cells. Instead, CRTC2 remains in the cell nucleus where it switches on a set of genes that make more proteins. This is important because:
- the production of these proteins distinguishes FLC from other types of liver cancers, namely hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC), and
- blocking CRTC2 function prevents the growth of FLC models.
CRTC2 works together with the p300 protein to regulate genes involved in cellular metabolism, growth, and inflammation. The researchers discovered that not only does SIK loss activate CRTC2 in DNAJB1-PRKACA expressing cells, it also increases p300 protein levels. Medicines against p300 are being tested against various cancers in clinical trials. Notably, treatment of the FLC models with a p300 inhibitor effectively suppressed tumor growth.
In summary, this work provides insights into the network by which DNAJB1-PRKACA causes cancer growth, revealing a central role of blocking SIKs and activating CRTC2 and p300. Fibrolamellar carcinoma currently lacks effective treatment options, particularly for patients with advanced or metastatic disease. Further studies of targeting p300 or CRTC2 or the development of approaches to restore the function of SIKs could lead to new treatments that may improve outcomes for patients with FLC. Another notable aspect of this study is that the pathways that cause this rare cancer overlap with those that are essential for more common types of pancreatic and lung cancers. By understanding these shared pathways, there may be opportunities to apply knowledge gained from each disease to guide treatments and preventive strategies across the different cancers.
The full text of the article can be accessed here.